Publications
Peer-reviewed publications in computational materials science and machine learning.
2025
- IEEE AESS
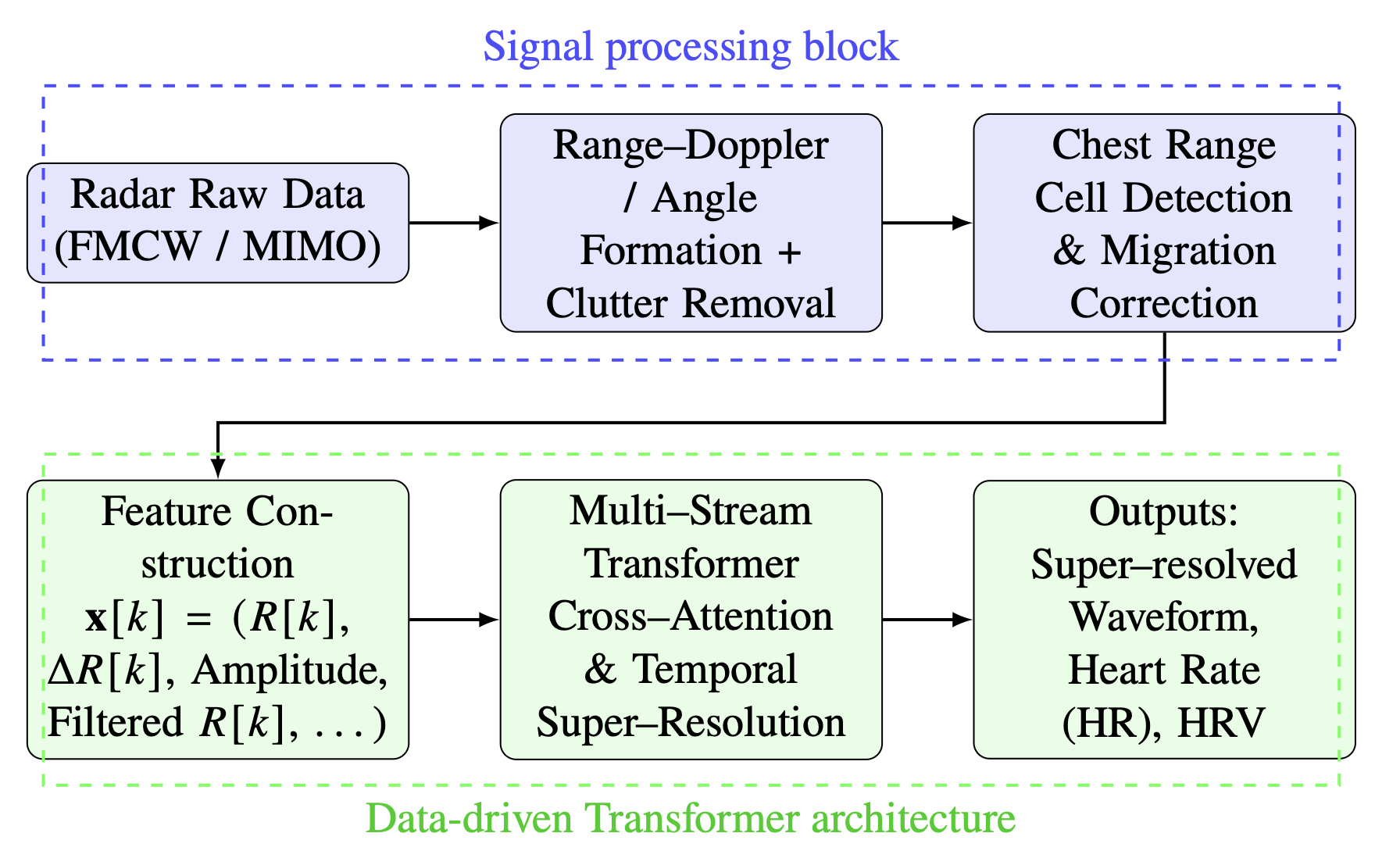 Physics-Guided Transformer Modeling of Radar-Based Heartbeat Monitoring in Dynamic ScenariosAmir Hosein Oveis, Mateo Pardi, Saba Kharabadze, and 1 more authorSep 2025Winning proposal for the 2026 IEEE AESS Heartbeat Challenge
Physics-Guided Transformer Modeling of Radar-Based Heartbeat Monitoring in Dynamic ScenariosAmir Hosein Oveis, Mateo Pardi, Saba Kharabadze, and 1 more authorSep 2025Winning proposal for the 2026 IEEE AESS Heartbeat ChallengeWinner
This proposal addresses the Radar Challenge on robust heartbeat monitoring in the presence of random body motion (RBM). Our approach improves the estimation of heartbeat and its variability by combining conventional radar signal processing techniques with data-driven machine learning methods. We construct a multidimensional time-series representation from the stabilized chest signal, which serves as an input to the Transformer model. In addition, we propose a novel data augmentation strategy, where RBMs are modeled and simulated with stochastic processes. This increases the diversity of the training data and helps the model adapt to uncontrolled real-world conditions. The main innovations compared with the existing literature are: (i) the physics-guided design of a meaningful time-series input for the Transformer, and (ii) a stochastic augmentation method for synthetic RBM signals. Together, these strategies aim to achieve robust and reliable radar-based heartbeat monitoring in dynamic scenarios.
@misc{kharabadze2025heartbeat, title = {Physics-Guided Transformer Modeling of Radar-Based Heartbeat Monitoring in Dynamic Scenarios}, author = {Oveis, Amir Hosein and Pardi, Mateo and Kharabadze, Saba and Kumar, Ajeet}, year = {2025}, month = sep, howpublished = {IEEE AESS Heartbeat of the Aerospace and Electronic Systems Group Challenge Winner}, note = {Winning proposal for the 2026 IEEE AESS Heartbeat Challenge}, }
2024
- PhD Thesis
 Machine Learning and Ab Initio Insights into the Design of Lithium-Based MaterialsSaba KharabadzePhD Thesis, 2024
Machine Learning and Ab Initio Insights into the Design of Lithium-Based MaterialsSaba KharabadzePhD Thesis, 2024Materials research, spanning physics, chemistry, and engineering, underpins technological innovation by enabling precise control of materials at the atomistic level. Nevertheless, the challenges of exploring the vast chemical space efficiently and cost-effectively remain. Quantum mechanical methods, such as density functional theory (DFT), while theoretically powerful, often prove computationally expensive for large systems. This thesis showcases the development of accurate machine learning potentials which aid in prediction of new materials. I have chosen \SKtwo main studies to highlight the work conducted during my PhD research. In the investigation of the Li-Sn binary system, which has been considered for energy storage applications, we focused on identifying new stable crystal structures. After constructing an interatomic description model for Li-Sn, we introduced a protocol for finding stable compounds at both low and high temperatures. Although this binary had been thoroughly explored in two recent ab initio predictive studies, we found two overlooked compounds thermodynamically stable at ambient conditions. Building on this proof-of-principle study, we expanded the scope of investigation to a much larger set of binaries to check the protocol’s general performance. The investigated M-Sn binaries, where M = Na, Ca, Cu, Pd, and Ag, have a potential for a wider range of applications in energy storage, electronics packaging, and superconductivity. The application of the developed approach has led to the identification of a large number of stable phases at various synthesis conditions. The findings of these two studies demonstrated a great promise, as the discovery of over 30 new stable crystal structures has dramatically increased the number of successful predictions based on machine learning potentials. The third study was dedicated to the analysis of thermodynamic stability of Li-B-C compounds, that had been attracting a lot of attention as potential record-breaking conventional superconductors. We utilized DFT to characterize both well-established and recently reported Li-B-C phases. Our work included demonstration and rationalization of inconsistencies in previously proposed crystal structure solutions. One of the main outcomes is the construction of the phase diagram of the LiBC delithiation. Our findings reveal an incomplete knowledge of the Li–B–C ternary and the need for further experimental exploration of this intriguing materials system.
@article{kharabadze2024thesis, title = {Machine Learning and Ab Initio Insights into the Design of Lithium-Based Materials}, author = {Kharabadze, Saba}, year = {2024}, publisher = {State University of New York at Binghamton}, doi = {https://www.proquest.com/openview/27b60b7af9e576913426999fff21eb47/1?pq-origsite=gscholar&cbl=18750&diss=y}, journal = {PhD Thesis} }
2023
- PCCP
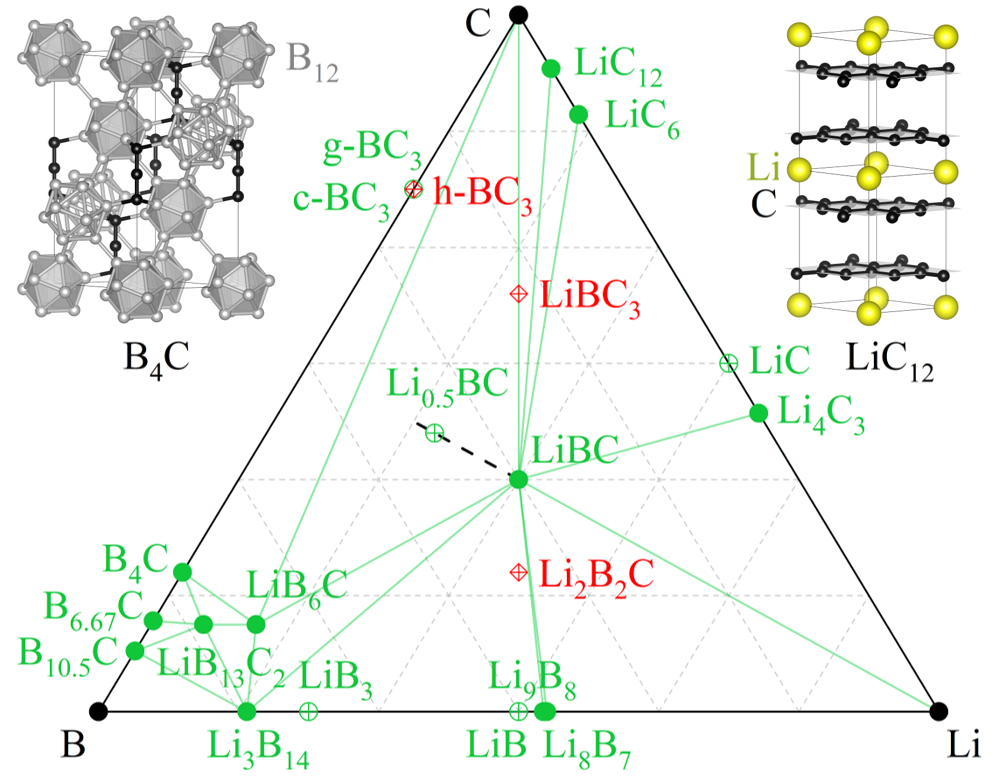 Thermodynamic stability of Li-B-C compounds from first principlesSaba Kharabadze, Matthew Meyers, Christopher R. Tomassetti, and 3 more authorsPhysical Chemistry Chemical Physics, 2023
Thermodynamic stability of Li-B-C compounds from first principlesSaba Kharabadze, Matthew Meyers, Christopher R. Tomassetti, and 3 more authorsPhysical Chemistry Chemical Physics, 2023Prediction of high-Tc superconductivity in hole-doped LixBC two decades ago has brought about an extensive effort to synthesize new materials with honeycomb B–C layers, but the thermodynamic stability of Li–B–C compounds remains largely unexplored. In this study, we use density functional theory to characterize well-established and recently reported Li–B–C phases. Our calculation of the Li chemical potential in LixBC helps estimate the (T,P) conditions required for delithiation of the LiBC parent material, while examination of B–C phases helps rationalize the observation of metastable BC3 polymorphs with honeycomb and diamond-like morphologies. At the same time, we demonstrate that recently reported BC3, LiBC3, and Li2B2C phases with new crystal structures are both dynamically and thermodynamically unstable. With a combination of evolutionary optimization and rational design, we identify considerably more natural and favorable Li2B2C configurations that, nevertheless, remain above the thermodynamic stability threshold.
@article{kharabadze2023libc, title = {Thermodynamic stability of Li-B-C compounds from first principles}, author = {Kharabadze, Saba and Meyers, Matthew and Tomassetti, Christopher R. and Margine, Elena R. and Mazin, Igor I. and Kolmogorov, Alexey N.}, journal = {Physical Chemistry Chemical Physics}, volume = {25}, pages = {7344--7353}, year = {2023}, publisher = {Royal Society of Chemistry}, doi = {https://doi.org/10.1039/D2CP05500G}, } - PCCP
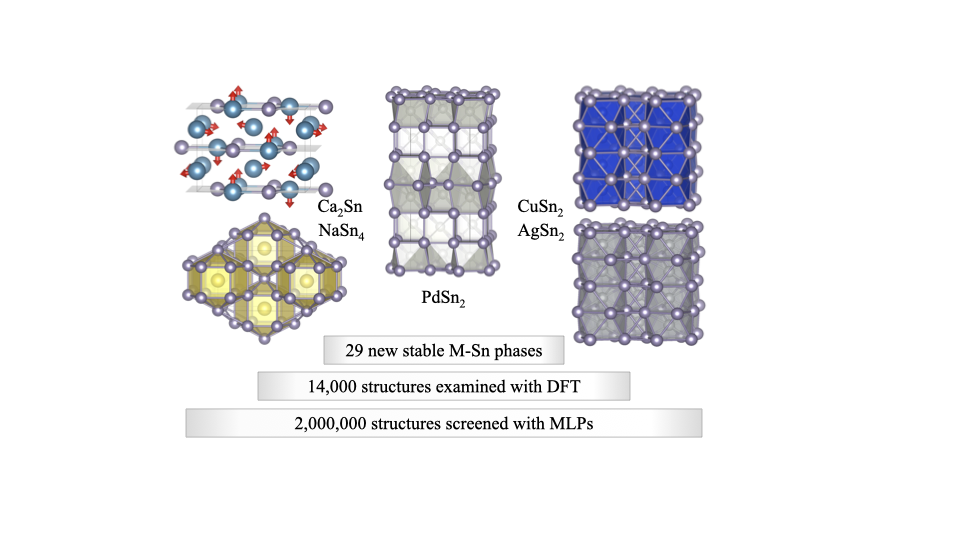 Machine learning search for stable binary Sn alloys with Na, Ca, Cu, Pd, and AgAidan Thorn, David Gochitashvili, Saba Kharabadze, and 1 more authorPhysical Chemistry Chemical Physics, 2023
Machine learning search for stable binary Sn alloys with Na, Ca, Cu, Pd, and AgAidan Thorn, David Gochitashvili, Saba Kharabadze, and 1 more authorPhysical Chemistry Chemical Physics, 2023We present our findings of a large-scale screening for new synthesizable materials in five M–Sn binaries, M = Na, Ca, Cu, Pd, and Ag. The focus on these systems was motivated by the known richness of M–Sn properties with potential applications in energy storage, electronics packaging, and superconductivity. For the systematic exploration of the large configuration space, we relied on our recently developed MAISE-NET framework that constructs accurate neural network interatomic potentials and utilizes them to accelerate ab initio global structure searches. The scan of over two million candidate phases at a fraction of the typical ab initio calculation cost has uncovered 29 possible intermetallics thermodynamically stable at different temperatures and pressures (1 bar and 20 GPa). Notable predictions of ambient-pressure materials include a simple hP6-NaSn2 phase, fcc-based Pd-rich alloys, tI36-PdSn2 with a new prototype, and several high-temperature Sn-rich ground states in the Na–Sn, Cu–Sn, and Ag–Sn systems. Our modeling work also involved ab initio (re)examination of previously observed M–Sn compounds that helped explain the entropy-driven stabilization of known Cu–Sn phases. The study demonstrates the benefits of guiding structure searches with machine learning potentials and significantly expands the number of predicted thermodynamically stable crystalline intermetallics achieved with this strategy so far.
@article{thorn2023sn, title = {Machine learning search for stable binary Sn alloys with Na, Ca, Cu, Pd, and Ag}, author = {Thorn, Aidan and Gochitashvili, David and Kharabadze, Saba and Kolmogorov, Alexey N.}, journal = {Physical Chemistry Chemical Physics}, volume = {25}, pages = {22415--22436}, year = {2023}, publisher = {Royal Society of Chemistry}, doi = {10.1039/D3CP02817H}, }
2022
- npj Comput. Mater.
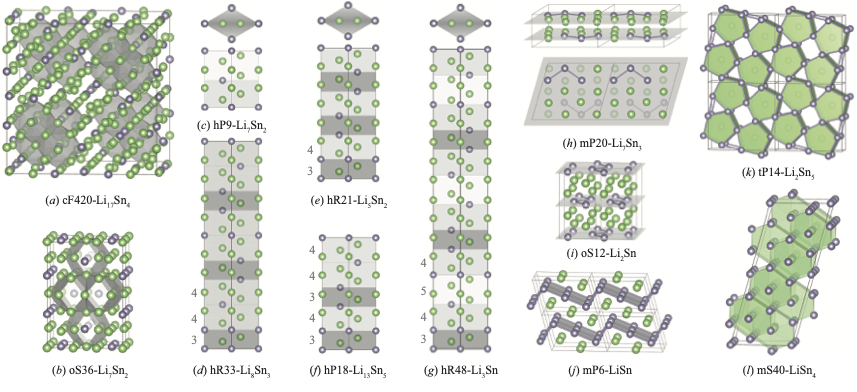 Prediction of stable Li-Sn compounds: boosting ab initio searches with neural network potentialsSaba Kharabadze, Aidan Thorn, Ekaterina A. Koulakova, and 1 more authornpj Computational Materials, 2022
Prediction of stable Li-Sn compounds: boosting ab initio searches with neural network potentialsSaba Kharabadze, Aidan Thorn, Ekaterina A. Koulakova, and 1 more authornpj Computational Materials, 2022The Li-Sn binary system has been the focus of extensive research because it features Li-rich alloys with potential applications as battery anodes. Our present re-examination of the binary system with a combination of machine learning and ab initio methods has allowed us to screen a vast configuration space and uncover a number of overlooked thermodynamically stable alloys. At ambient pressure, our evolutionary searches identified an additional stable Li3Sn phase with a large BCC-based hR48 structure and a possible high-T LiSn4 ground state. By building a simple model for the observed and predicted Li-Sn BCC alloys we constructed an even larger viable hR75 structure at an exotic 19:6 stoichiometry. At 20 GPa, low-symmetry 11:2, 5:1, and 9:2 phases found with our global searches destabilize previously proposed phases with high Li content. The findings showcase the appreciable promise machine-learning interatomic potentials hold for accelerating ab initio prediction of complex materials.
@article{kharabadze2022lisn, title = {Prediction of stable Li-Sn compounds: boosting ab initio searches with neural network potentials}, author = {Kharabadze, Saba and Thorn, Aidan and Koulakova, Ekaterina A. and Kolmogorov, Alexey N.}, journal = {npj Computational Materials}, volume = {8}, pages = {136}, year = {2022}, publisher = {Nature Publishing Group}, doi = {https://doi.org/10.1038/s41524-022-00825-4}, } - J. Phys. Chem. C
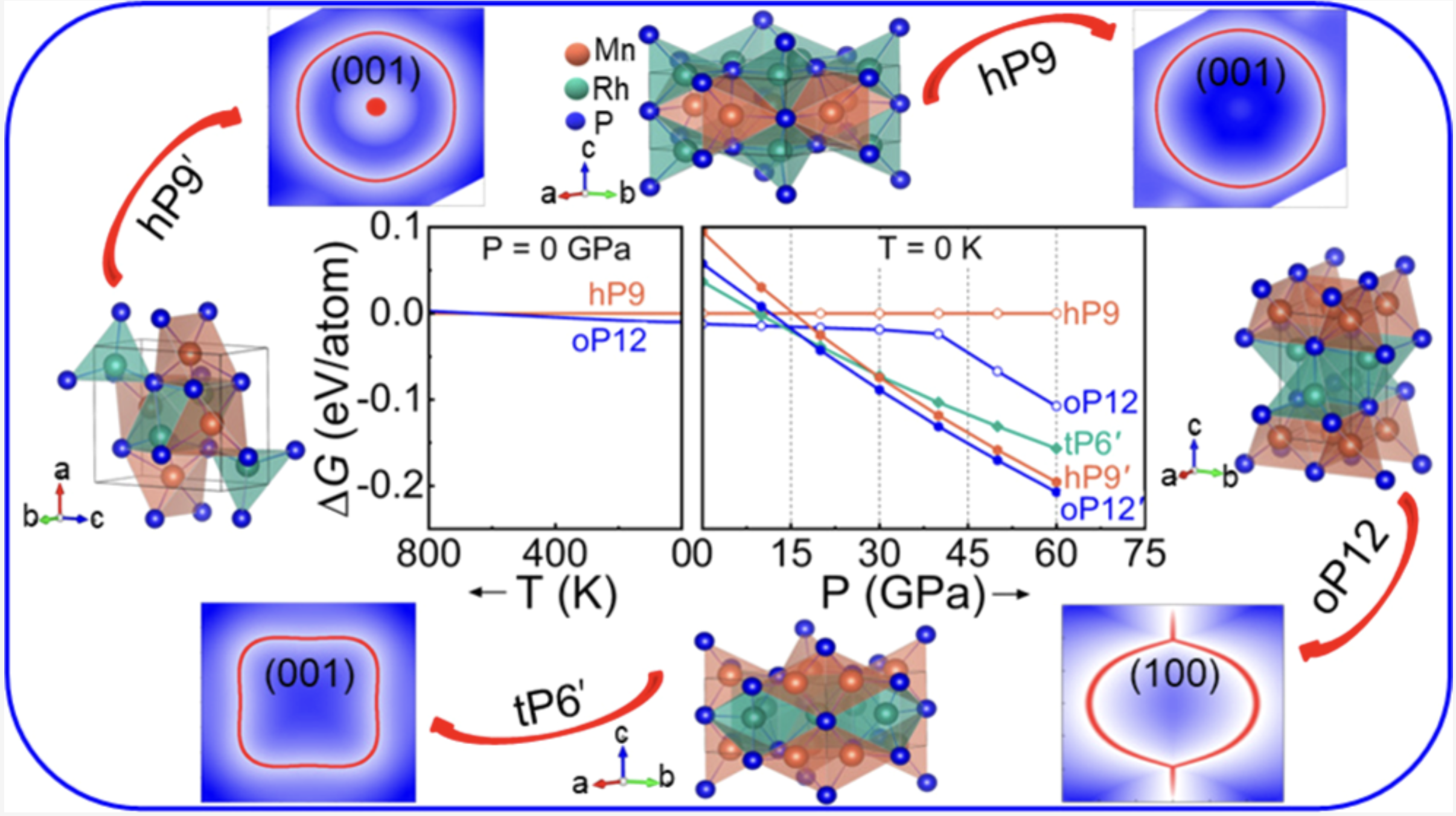 Prediction of Ground State Structures and Robust Weyl Fermionic States in MnRhPJaskirat Singh, A. Behatha, Saba Kharabadze, and 3 more authorsThe Journal of Physical Chemistry C, 2022
Prediction of Ground State Structures and Robust Weyl Fermionic States in MnRhPJaskirat Singh, A. Behatha, Saba Kharabadze, and 3 more authorsThe Journal of Physical Chemistry C, 2022Topological metals are a new class of materials that feature Fermionic quasiparticles with the presence of non-trivial band crossings near the Fermi level. In this work, we focus on establishing crystal structure ground states and the corresponding topological properties of MnRhP. Under ambient pressure and low temperatures, we find that an orthorhombic oP12 polymorph is favored over the known hexagonal hP9 phase. Pressures above 15 GPa stabilize tetragonal (tP6′), hexagonal (hP9′), and orthorhombic (oP12′) phases with inverted population of metal sites. While oP12′ has the lowest enthalpy, we show that hP9′ is more consistent with the previous X-ray diffraction data collected at 60 GPa. Our analysis of hP9 and oP12 topological properties reveals the existence of nodal lines around the Γ-point that are gapped out when spin–orbit coupling effects are included and transform into Weyl nodes with opposite chirality near the Fermi level. The calculated large values of the anomalous Hall conductivity in hP9, oP12, and tP6′ and the Z2 topological invariant in the non-magnetic hP9′ can be used to verify the predicted non-trivial robust topological features of MnRhP under ambient and high pressures.
@article{singh2022mnrhp, title = {Prediction of Ground State Structures and Robust Weyl Fermionic States in MnRhP}, author = {Singh, Jaskirat and Behatha, A. and Kharabadze, Saba and Kolmogorov, Alexey N. and Vaitheeswaran, G. and Kanchana, V.}, journal = {The Journal of Physical Chemistry C}, volume = {126}, number = {40}, pages = {17328--17337}, year = {2022}, publisher = {American Chemical Society}, doi = {https://doi.org/10.1021/acs.jpcc.2c04603}, }
2021
- CPC
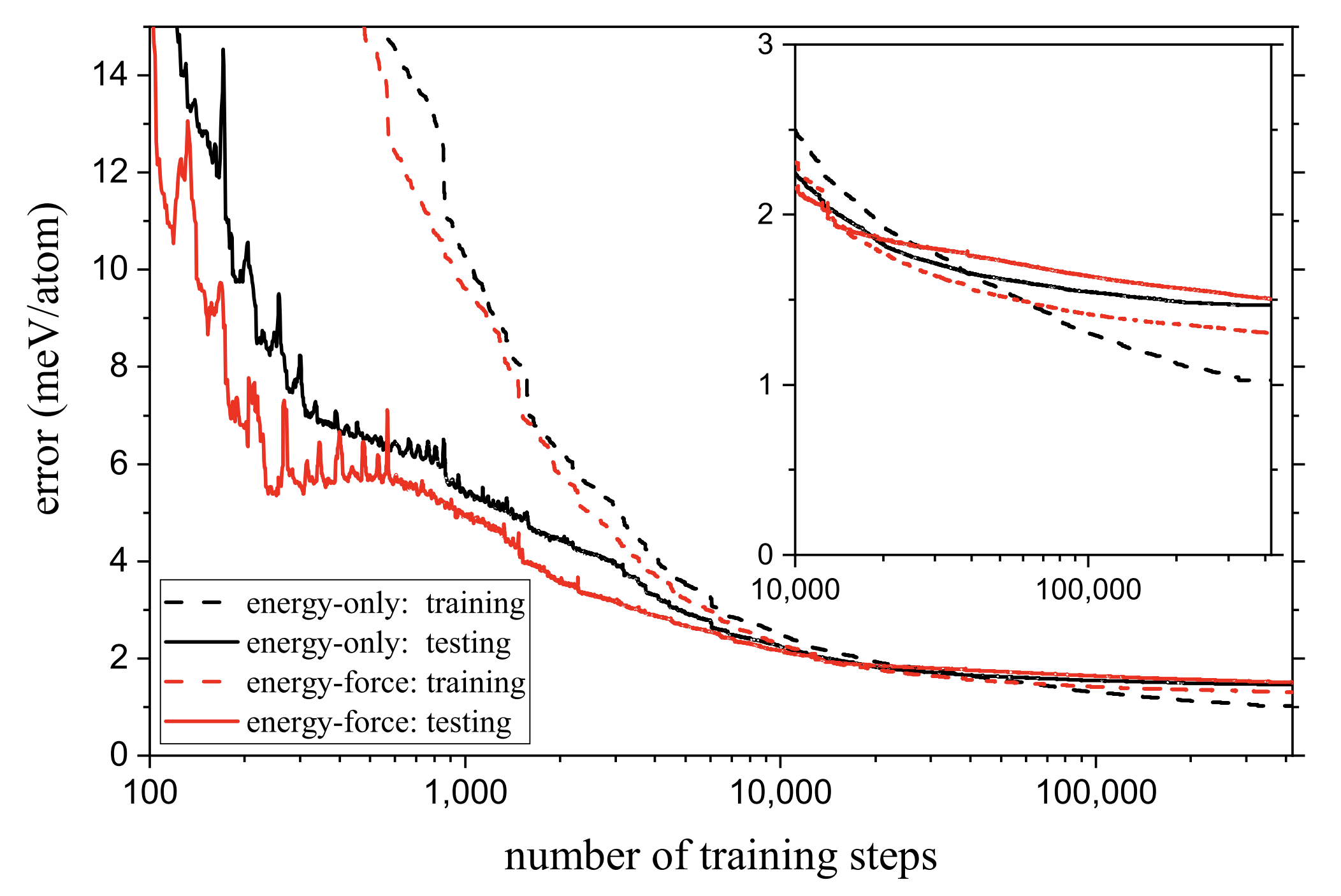 MAISE: Construction of neural network interatomic models and evolutionary structure optimizationSamad Hajinazar, Aidan Thorn, Ernesto D. Sandoval, and 2 more authorsComputer Physics Communications, 2021
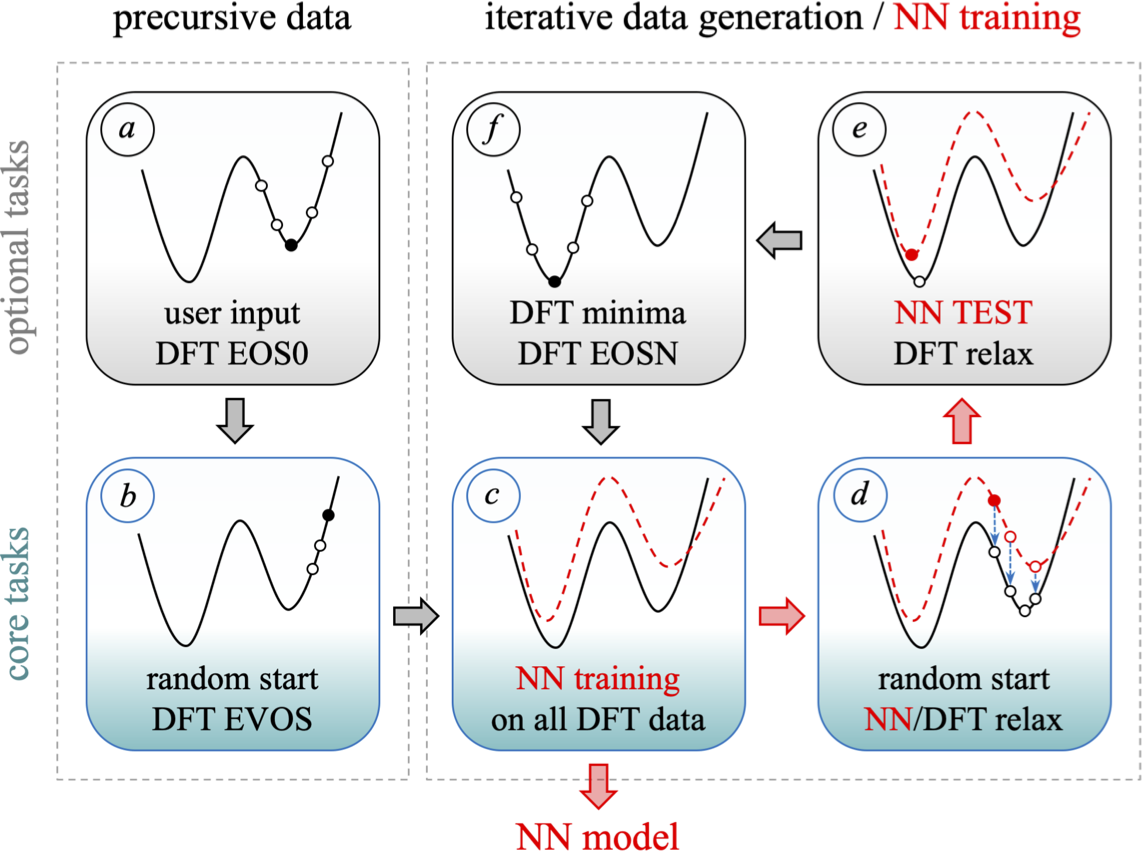
MAISE: Construction of neural network interatomic models and evolutionary structure optimizationSamad Hajinazar, Aidan Thorn, Ernesto D. Sandoval, and 2 more authorsComputer Physics Communications, 2021Module for ab initio structure evolution (MAISE) is an open-source package for materials modeling and prediction. The code’s main feature is an automated generation of neural network (NN) interatomic potentials for use in global structure searches. The systematic construction of Behler–Parrinello-type NN models approximating ab initio energy and forces relies on two approaches introduced in our recent studies. An evolutionary sampling scheme for generating reference structures improves the NNs’ mapping of regions visited in unconstrained searches, while a stratified training approach enables the creation of standardized NN models for multiple elements. A more flexible NN architecture proposed here expands the applicability of the stratified scheme for an arbitrary number of elements. The full workflow in the NN development is managed with a customizable ‘MAISE-NET’ wrapper written in Python. The global structure optimization capability in MAISE is based on an evolutionary algorithm applicable for nanoparticles, films, and bulk crystals. A multitribe extension of the algorithm allows for an efficient simultaneous optimization of nanoparticles in a given size range. Implemented structure analysis functions include fingerprinting with radial distribution functions and finding space groups with the SPGLIB tool. This work overviews MAISE’s available features, constructed models, and confirmed predictions.
@article{hajinazar2021maise, title = {MAISE: Construction of neural network interatomic models and evolutionary structure optimization}, author = {Hajinazar, Samad and Thorn, Aidan and Sandoval, Ernesto D. and Kharabadze, Saba and Kolmogorov, Alexey N.}, journal = {Computer Physics Communications}, volume = {259}, pages = {107679}, year = {2021}, publisher = {Elsevier}, doi = {10.1016/j.cpc.2020.107679}, }